SNPs, ChIPs and RNA
Attempts to understand complex phenotypes
Peter Humburg
15th April 2015
Overview
Variant annotation
- Variant annotations depend on quality of transcript annotations.
- Different annotation software may give different results.

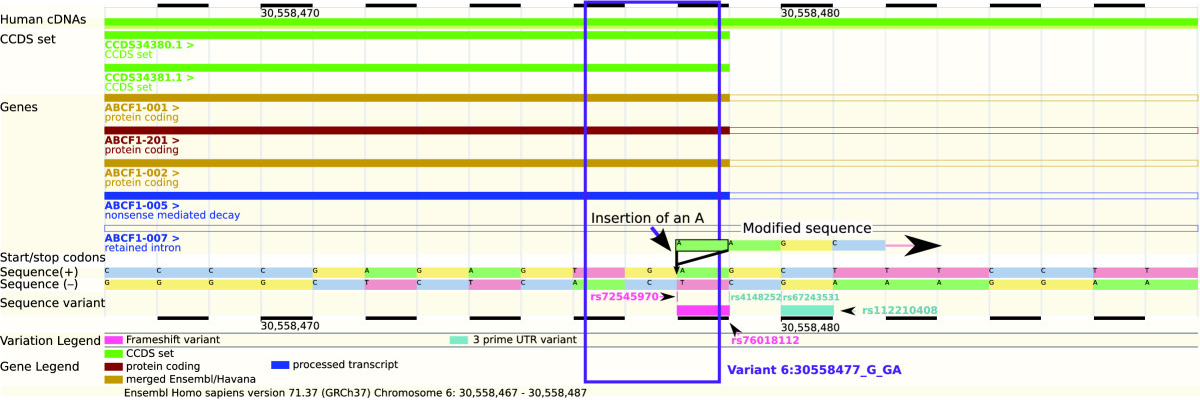
Results
- Identified genes enriched for PTVs.
- Strongest PTV enrichment observed for PPM1D.
- Observed clustering of variants in final exon.
Sequenced affected exon in 7,781 cases and 5,861 controls
- 18 PTVs in 6,912 breast cancer cases
- 12 PTVs in 1,121 ovarian cancer cases
- 1 PTV in controls
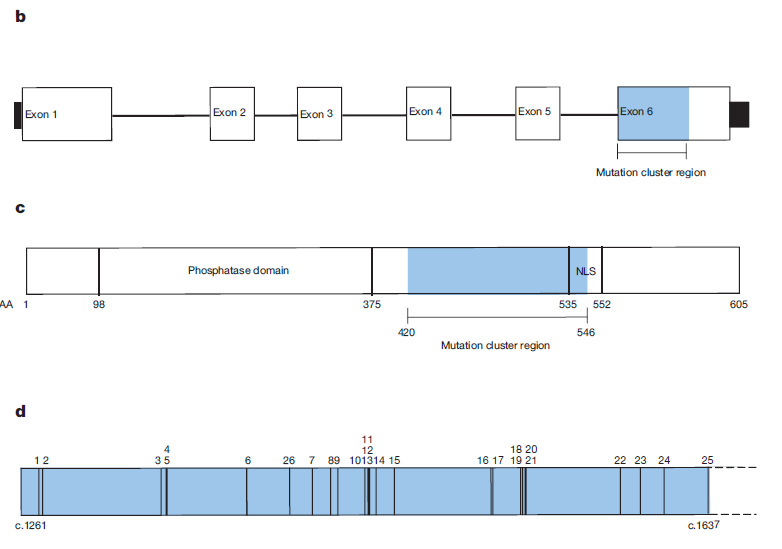
PPM1D
- Induced in p53 dependent manner
- Contributes to deactivation of p53
- Part of negative feedback loop required to escape p53-dependent cell cycle arrest
- Truncated proteins show increased activity.
- PPM1D over-expression previously associated with breast cancer.

Interpreting non-coding variants
- Impact of non-coding variants unclear
- Affected genes not obvious
- Effect on gene expression typically unknown
- Existing epigenetic data may help
- eQTL analyses can help to establish links between SNPs and genes


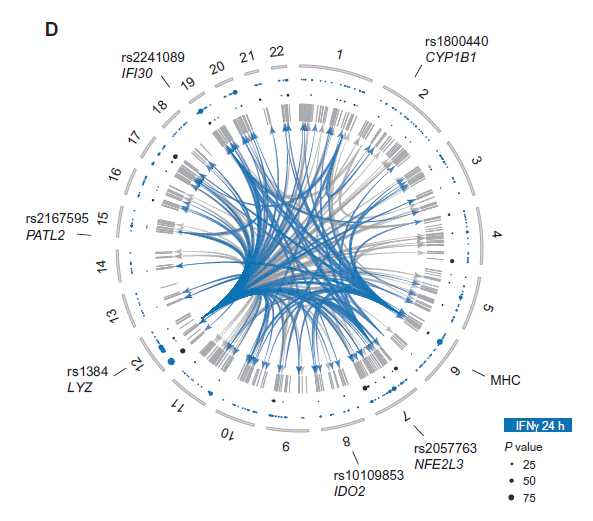
Using additional genome-wide data
ChIP-seq and RNA-seq data provide insight into the functional implications of genotypes. But need to consider
- Relevant cell type
- Relevant conditions


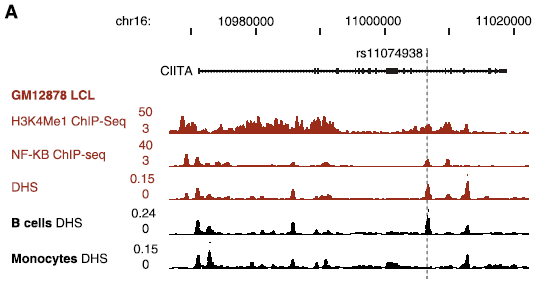
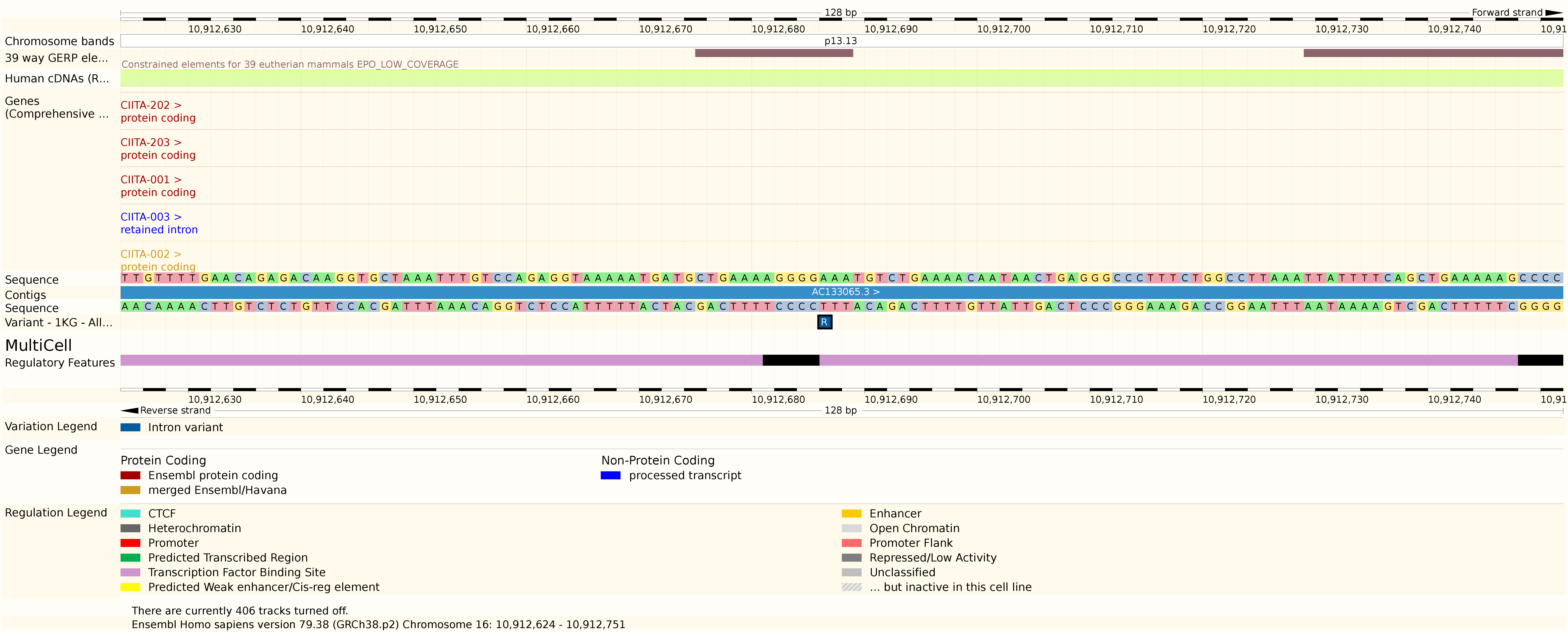
The role of rs11074938
- SNP (A/G) located in intron of CIITA
- Inside regulatory region
- ENCODE data shows DHS and NF-\(\kappa\)B binding
- CIITA is important regulator of MHC class II expression
- Could have implications for immune related diseases
(if this SNP affects CIITA)

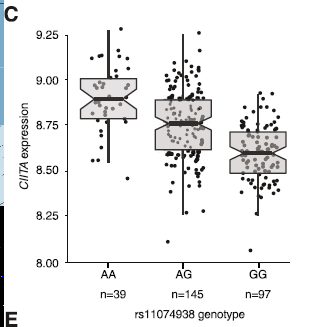
Changes to gene expression
- eQTL analysis in B cells shows reduced expression of CIITA associated with G allele
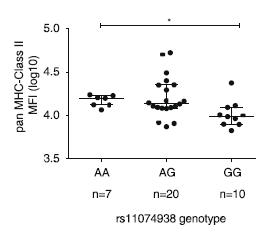
- Evidence for changes in expression of CIITA target genes
- Reduced presence of MHC class II proteins on cell surface associated with G allele
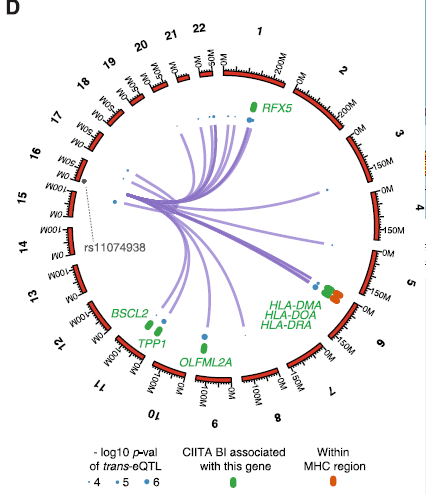
Transcription factor binding
- Reduced NF-\(\kappa\)B binding due to sequence change plausible
- Used existing ChIP-seq data\(^*\) to assess allele specific binding
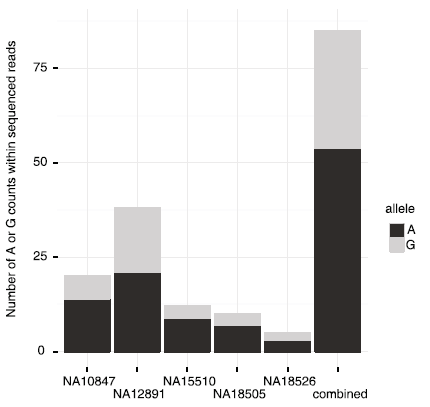
\(^*\) Kasowski et al., Science (2010). PMID: 20299548
Acknowledgments
Breast cancer risk and variant annotation
 |
Peter Donnelly |  |
Nazneen Rahman |
 |
Manuel A. Rivas |  |
Katie Snape |
 |
Andrew Rimmer | Elise Ruark | |
 |
Davis McCarthy |
Acknowledgments
eQTL and CIITA
 |
Julian C. Knight |
 |
Daniel Wong |
| Wanseon Lee | |
 |
Benjamin P. Fairfax |
 |
Seiko Makino |
